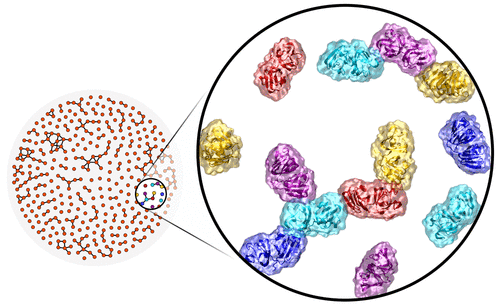
Protein aggregation is a process of critical importance to diseases ranging from Alzheimer's to cataract, but modeling it poses significant computational challenges. A new paper by the Butts and Tobias groups in the Journal of Physical Chemistry shows a new way of scaling up detailed, atomistic models of aggregation to much larger systems. By representing protein aggregates as tangled networks of monomers, the researchers are able to use statistical network models inspired by human interactions to reproduce many of the aggregates'
physical properties - and to do so on a much larger scale than could be achieved with existing methods. By combining detailed but expensive atomistic models with efficient, coarse-grained models, this works suggests a promising direction for probing the behavior of complex molecular systems with important implications for human health.